
PhyloWS BioHackathon Report
At present there is no standard web-service API for phylogenetic data that would allow integration of phylogenetic data and service providers into the programmable web. Hence, current approaches to integrate data and services into workflows are highly specific to the integration platform (CIPRES, Bioperl, Bio::Phylo, Kepler), and nearly unusable in other environments. This work group was formed to remedy this.
To begin with, we discussed what our scope for now would be. We accumulated use cases and requirements, proposed a simple API and started reference implementations. In addition, we had discussions over node metadata and the Operational Taxonomic Unit concept.
Node metadata
We need to be able to associate (typed) data with tree branches - the obvious example are branch lengths - but we usually also have (possibly multiple) support values associated with tree branches.
There is an extensive number of metadata elements one might want to record for nodes in a tree. Beyond the basic gene name, sequence ID, and taxon, metadata range from biogeography (area, lat/long), to host species (for analyses of host/parasite co-evolution), to functional attributes (GO terms, expression, subcellular location).
Some of these attributes may blur the line to actual character data for nodes (whether tips or internal). In fact, the notion of strictly distinguishing between node metadata and node character data may not be tenable or useful in reality; data may be metadata in one situation, and used as correlated character in another situation, and used for tree inference in a third situation.
There are essentially two ways to represent node metadata:
- Using a metadata schema (for example, an XML Schema, or a XML document definition, similar to metadata schemas in use in the library sciences such as DC, MARC21, etc).
- Pros: provides a clear expectation for clients as to what metadata to expect, and the meaning of those elements can be (hard-) coded into applications.
- Cons: limited to whatever the metadata schema allows for, and any new metadata elements deemed useful by emerging research cannot be readily expressed (and rectifying that requires a metadata schema update, which in turn affects client applications rather directly). In addition, the meaning of metadata elements cannot be dynamically inferred or stated; it is implicit from the XML DTD or Schema definition.
- Using attribute/value pairs, where attribute names are drawn from a controlled vocabulary.
- Pros: allows for an unlimited number of metadata elements, and easy representation of new elements as necessitated by new research approaches. The semantics of a metadata element can be as richly represented as desired, in a readily machine-readable form, by relating the attribute term to other ontology terms. The metadata representation is readily amenable to an OWL or RDF representation.
- Cons: a client cannot know in advance which metadata elements it may encounter (though it may limit itself to only interpret a pre-defined number of them), and the full semantics of a metadata element may change solely by the underlying ontology changing.
We feel that only using attribute/value pairs fits our and research requirements.
Operational Taxonomic Unit
The OTU perspective is an important use-case:
- Species tree hypothesis testing: splitting a given set of trees into subsets of trees as a function of compatibility to a given (set of) species tree(s). Degree of compatibility can be expressed as minimal sum of duplications needed to reconcile the gene with a species tree. I.e. measurement of the percentage of gene trees supporting an ecdysozoan versus a coelomata hypothesis.
- Problem: the query topology will be given with either gene name labels, or species name labels, but the labels of the trees will be OTUs.
- Hence, each OTU needs to be linked to the gene name(s) and taxon names, and it needs to be possible to specify that matching tree nodes use the linked taxon or gene names.
- The analysis mentioned above could be extended by asking questions about the (majority of) functional categories supporting a given species tree. These examples require association of the following data with gene tree nodes: taxonomy identifier, sequence identifier (which then, ideally, allows to retrieve functional data, such as GO).
- Gene tree analysis: similar to the Zmasek et al (2007) paper, one may want to build alignments and phylogenetic trees for all members of each protein (family) of a biological network (e.g. apoptsis). After loading the trees into a database, one could then query the database for those gene trees that exhibit a given pattern (e.g. lineage specific gene expansion or gene loss).
- In molecular and comparative genomics applications, one may want to find all trees that have been built for a certain sequence.
- Problem: As above, querying by sequence will give the gene name or the sequence accession number to match by, but tree nodes will have OTUs as labels.
- We discussed whether we need identifiers for OTUs.
- Pros: Rather than many individual idiosyncratic schemes for encoding sequence ID and taxon (and possibly additional information) into an OTU label, a single identifier could be resolved to the metadata using a common mechanism (such as LSID). Alternatively, one could standardize on a common encoding mechanism, that could then be parsed by a common mechanism.
- Cons: If using an (presumably opaque) identifier for OTUs, one ought to be able to expect that the same combination of sequence ID, taxon name (where one often implies the other, unless sequence ID is really an ambiguous gene name), and additional metadata (such as allele, population sample, etc) results in the same identifier, in essence necessitating an OTU identifier registry, or a common algorithm for constructing the identifier (which would then no longer be opaque). A standardized encoding mechanism would need to be widely supported and adopted.
Formalizing scope, use cases, requirements (Tuesday)
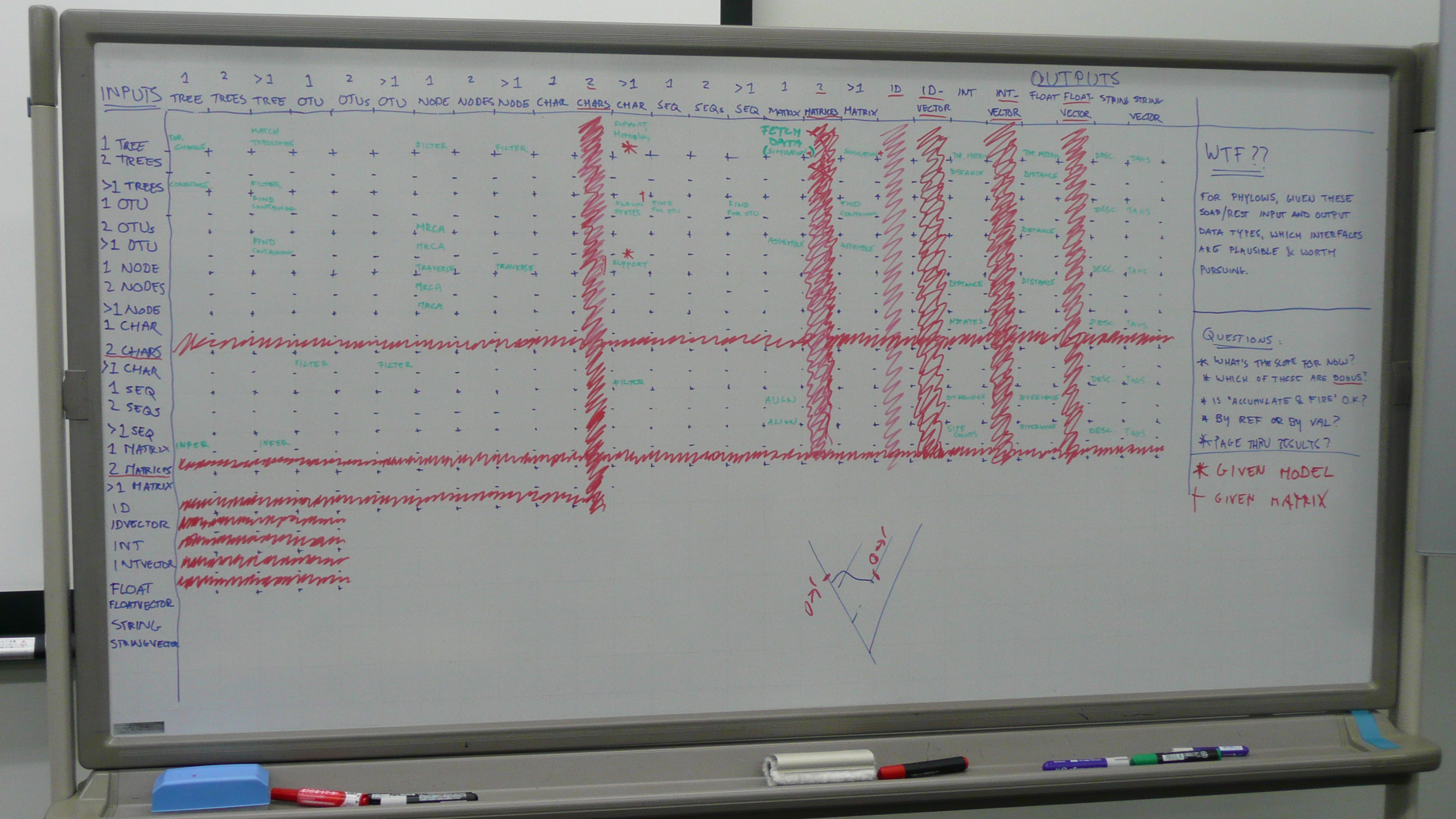
In a white board exercise the next day, we identified plausible input and output data types for phyloinformatic webservices. Plausibility is defined by our being able to imagine use cases (no time line for implementation implied, the goal here is to come up with interfaces)
- Inputs - The input data types defined here do not imply pass-by-value, i.e. "One Tree" could be some kind of identifier. In addition, it looks like several services need multiple data types (e.g. a matrix + a tree, in order to return a tree score). Given that accumulate-and-fire (i.e. stateful operations, where you first post a matrix, then a tree) is a Bad Thing, and that tunneling multiple data types in a single attachment or body (e.g. a chunk of nexus/nexml) is also a Bad Thing, it seems that these combinations of data would be submitted as multiple post parameters.
- One Tree - exactly one tree, which might function as a query topology, as an input for topology metric calculations, or as something for which associated data (matrices) and metadata might be retrieved
- Pair of Trees - exactly two trees, which function as inputs for tree reconciliation (e.g. duplication inference) or for tree-to-tree distance calculations
- Set of Trees - input for consensus calculations, or as query topologies
- One OTU - exactly one OTU for which associated data (trees or matrices that contain it) and metadata might be retrieved
- Pair of OTUs - exactly two OTUs, as input for topological queries (MRCA) and calculations (patristic distance)
- Set of OTUs - input for topological queries (MRCA) and for which data (trees or matrices that contain them) and metadata might be retrieved
- One Node - input for tree traversal operations (parent, children) and for which metadata might be retrieved
- Pair of Nodes - input for topological queries (MRCA) and calculations (patristic distance)
- Set of Nodes - input for topological queries (MRCA)
- One Character - exactly one character (matrix column) for which calculations are performed (variability) and metadata is retrieved
- Set of Characters - input as filter predicate, to retrieve OTUs that contain recorded states for the characters
- One Character State Sequence - for which metadata is retrieved
- Pair of Character State Sequences - as input for pairwise alignments, as input to calculate pairwise divergence
- Set of Character State Sequences - as input for multiple sequence alignment
- Character State Matrix - as input for inference (of one tree or set of trees), as input for calculations (average sequence divergence) and for which metadata is retrieved
- Outputs - In addition to the mirroring the inputs described above, some 'primitives' may be required:
- Int - an integer, for things such as topology metrics (node counts) tree-to-tree distances (in branch moves) node distances (in number of nodes in between), character state counts, sequence divergence (substitution counts, site counts)
- Float - a floating point value, for topology metrics (balance, stemminess, resolution) tree-to-tree distances (symmetric difference), patristic distance, sequence divergence
- String - for metadata, e.g. descriptions
- Stringvector - for metadata, e.g. a set of tags
For these I/O combinations, we tried to imagine a use case, and moved these to NESCent wiki. We classified each use case according to their scope and requirements, which yielded the following three main scopes of interest to implement:
- Phylogenetic tree database, i.e. a persistence service based on BioSQL
- Phylogenetic data conversion, i.e. make sure that inputs and outputs of data work syntactically
- Phylogenetic data analysis, i.e. actual computations
API design
Assuming that syntax and semantics of the data can be solved, what would be the syntax and semantics of making the connections?
- API design principles - by the end of the hackathon, we had identified the following RESTful API principles, but they will continue to be developed elsewhere:
- Stateless REST, no accumulate-and-fire
- Architecture modeled as resources on which CRUD operations are performed
- Use common HTTP methods (GET, PUT, POST, DELETE)
- Describe RESTful API in WSDL 2.0
- Reuse prior art
- URL API - We started to design a URL API (pathinfo + query strings) on the NESCent Wiki
Actual implementation
All displacement activities aside, we did some coding:
- Started conversion and persistence services
- Improve plumbing (format support, database support)
Moving forward
- A web-services specification is being developed, initially based on RESTful interfaces. We also intend to create a WSDL 2.0 compliant service description, which can have alternative service bindings to REST (HTTP binding) or SOAP (SOAP binding). If possible, we intend to make the query service SRU v1.2 compliant.
- Implement services.
Attachments
-
P1070128.JPG
 (2.2 MB) - added by rvos
17 years ago.
(2.2 MB) - added by rvos
17 years ago.
{kind=link}